
Professional Paper Writing Service
An essay paper writing service that is dedicated to providing papers of the highest quality within the given deadlines.


100% Original Papers
We guarantee that your personal essay writer is giving you a final product that is 100% original, whether it's a resume, a term paper, a business plan, or an essay.
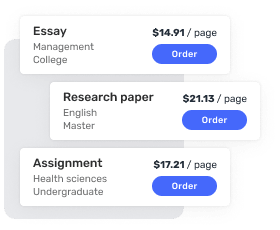
Affordable & Flexible Pricing
There are plenty of cheap essay writing services online. However, many essay writers do not deliver quality papers. Highly-qualified personal essay writer starting at just $10 per page

Privacy & Confidentiality
Your right to privacy is something we don’t take lightly. That's why we offer guaranteed confidential service. You can have complete faith that whatever sort of paper writing help you need

24/7 Customer Support Team
We know you may have questions throughout the process. Therefore, our customer support team is available 24/7 to answer your questions.
Hire Professional Writers
When you hire a personal essay writer, it is of the utmost importance that you are confident in the final product. service, ready to meet your writing needs.
When I received a paper revision from my new college professor, I was desperate as I didn’t know what to do.
You people have been really patient with me as I created mechanical engineering content. It’s not an easy topic to handle, yet I can say that I worked with a skilled specialist.
When I received a paper revision from my new college professor, I was desperate as I didn’t know what to do.
When I received a paper revision from my new college professor, I was desperate as I didn’t know what to do. If not for Academized, I would be doomed.
Paper Writing Help Online
Do you need help writing a paper? Don't know where to start, or just don't feel like you have the time to tackle the job? Or maybe you do have the time…but you can think of better ways to use it!
Active Writers
Completed Orders
Frequently Asked Questions
How do I place an order with you
All you need to do is enter your paper guidelines by filling out forms that are quick and simple. Then, our team will assign you a personal essay writer most suitable to handle your task. You’ll receive a message and an email when the writer is assigned and then when it is time for you to download papers.
How much does it cost to hire writer
You may think you need to spend a lot of money to hire a writer for an essay. However, you can get paper writing help starting at just $10 per page. The price for essay writing help will depend on the deadline, academic level, and the number of pages. The essay calculator on the main page will give you an estimate of the total cost for your paper. Moreover, we regularly offer discounts and send out promos weekly, keeping paper writing help affordable.
Do I need to pay you upfront
Yes, our service is pre-paid. If you want to make sure we have a professional writer specifically for your project before paying, try out our “Free Inquiry” option. It will allow you to make the payment only after you are connected to a suitable writer and have made all the necessary clarifications with them.
How fast can you complete an essay?
The expert essay writer can take on your assignment as soon as you submit the paper guidelines and payment. We know that you may need help writing a paper quickly. Our assignment writers can get your essay written in as little as 6 hours.
Will the writer follow my requirements?
Yes. Your personal essay writer will carefully review your paper guidelines. The more information you can provide when you hire a writer for an essay, the better. We encourage you to upload rubrics and other helpful files and indicate any other preferences in the paper details of the order form
Who are the writers?
We hire assignment writers from all over the world based on their skills, knowledge, and experience. English is a native language for most of our expert essay writers, but we make sure that all of them are fluent.