Nursing Paper Example on Ehlers-Danlos Syndrome
(Nursing Paper Example on Ehlers-Danlos Syndrome) Ehlers-Danlos Syndrome (EDS) is a group of rare, inherited connective tissue disorders that affect the skin, joints, and blood vessels. It is characterized by a range of symptoms, including hypermobility of joints, skin hyperextensibility, and tissue fragility, which vary in severity depending on the subtype. EDS is primarily caused by genetic mutations affecting the structure or synthesis of collagen, a vital protein in connective tissue. These mutations lead to compromised tissue integrity and function, resulting in complications such as chronic pain, joint instability, and, in severe cases, vascular rupture. While there are 13 recognized subtypes of EDS, each linked to distinct genetic abnormalities, the syndrome’s diagnosis and management remain challenging due to its diverse presentation.
Causes of Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome (EDS) is caused by genetic mutations that affect connective tissue components, primarily collagen. Collagen is a crucial protein that provides strength and elasticity to tissues such as skin, ligaments, and blood vessels. Mutations that impair collagen production or structure lead to weakened connective tissues, which underlie the manifestations of EDS.
Genetic Mutations and Subtypes
There are 13 recognized subtypes of EDS, each linked to specific genetic mutations. For example, classical EDS results from mutations in the COL5A1 and COL5A2 genes, which encode type V collagen. These mutations lead to fragile and hyperextensible skin. In vascular EDS, mutations in the COL3A1 gene disrupt the synthesis of type III collagen, which compromises blood vessel integrity and increases the risk of vascular rupture. Other subtypes, such as hypermobile EDS, lack a clearly defined genetic mutation, although recent studies suggest potential links to the TNXB gene.
Hereditary Patterns
The mode of inheritance varies by subtype. Autosomal dominant inheritance is observed in most forms, including classical and vascular EDS, meaning an affected individual has a 50% chance of passing the condition to their offspring. Autosomal recessive inheritance is less common and is seen in subtypes like dermatosparaxis EDS, requiring both parents to carry a copy of the mutated gene.
Environmental and Epigenetic Influences
While EDS is predominantly genetic, environmental and epigenetic factors may influence the severity of symptoms. Factors such as mechanical stress on joints, repetitive strain, or nutritional deficiencies can exacerbate symptoms but do not directly cause the condition.
Signs and Symptoms of Ehlers-Danlos Syndrome
The signs and symptoms of Ehlers-Danlos Syndrome (EDS) can vary widely depending on the subtype, but they primarily affect the skin, joints, and blood vessels. Common symptoms include hypermobility of joints, fragile skin, and a tendency to bruise easily.
Skin Involvement
One of the hallmark signs of EDS is hyperelasticity, or the ability of the skin to stretch beyond normal limits. Skin in individuals with EDS may be soft, velvety, and fragile, leading to frequent bruising and scarring. These individuals often develop atrophic scars that are thin, narrow, and stretch easily. In classical EDS, these skin changes are more pronounced, with a higher risk of wound complications and delayed wound healing.
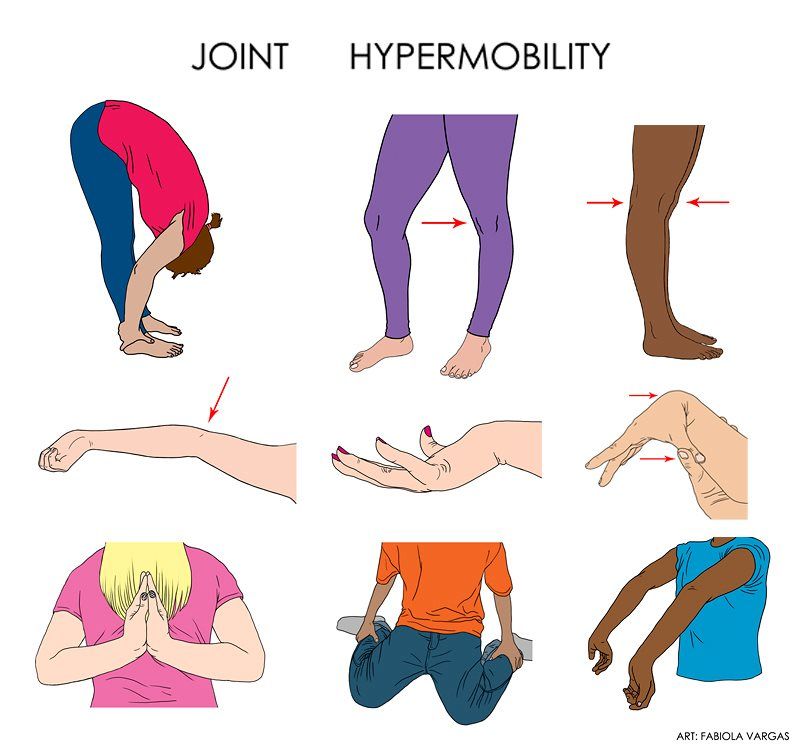
Joint Hypermobility
Joint hypermobility is another defining feature, particularly in the hypermobile subtype of EDS. Affected individuals may experience loose or unstable joints that easily dislocate or subluxate. This hypermobility can lead to chronic joint pain, instability, and an increased risk of injury. Joint hypermobility often affects the fingers, knees, elbows, and shoulders.
Vascular Complications
In more severe forms of EDS, such as vascular EDS, blood vessels, internal organs, and tissues are more fragile. This increases the risk of spontaneous ruptures, particularly in the arteries, which can be life-threatening. Individuals with vascular EDS may experience internal bleeding, organ rupture, or arterial dissection, which can cause severe complications.
Other Symptoms
Additional symptoms may include muscle weakness, scoliosis, and early onset of osteoarthritis due to joint instability. Individuals with EDS often experience chronic fatigue, and in some cases, there may be a higher susceptibility to developing cardiac issues like mitral valve prolapse.
These symptoms vary in severity and can significantly impact daily functioning, making early diagnosis and intervention important for managing the condition.
Pathophysiology of Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome (EDS) is primarily caused by genetic mutations that affect the production or processing of collagen, a key structural protein in the body. Collagen provides strength and elasticity to connective tissues, and its defects result in the hallmark features of EDS, such as hyper-elastic skin and joint hypermobility.
Collagen Deficiency or Abnormalities
Collagen plays a crucial role in the structural integrity of skin, tendons, ligaments, and blood vessels. In EDS, mutations in genes that encode collagen molecules, or in genes responsible for collagen processing enzymes, lead to weakened collagen fibers. In classical EDS, for instance, mutations affect type I or type III collagen, making the collagen fibers weaker and more prone to stretching. In vascular EDS, mutations in the COL3A1 gene affect type III collagen, leading to fragile blood vessels and internal organs, which increases the risk of life-threatening ruptures.
Impact on Connective Tissue
Collagen abnormalities disrupt the normal function of connective tissues, impairing their ability to support and protect internal structures. As a result, individuals with EDS experience increased skin fragility, joint hypermobility, and soft tissue that is easily injured. The instability of collagenous structures also contributes to the characteristic pain, instability, and frequent dislocations seen in affected individuals.
Increased Risk of Vascular and Organ Rupture
In severe forms like vascular EDS, the defective collagen in the blood vessels leads to their fragility, making them more susceptible to rupture. This defect also affects internal organs such as the intestines and the uterus, increasing the risk of spontaneous organ rupture and internal bleeding. The pathophysiology of EDS thus underscores the essential role of collagen in maintaining tissue integrity, with disruptions leading to widespread systemic issues.
(Nursing Paper Example on Ehlers-Danlos Syndrome)
Diagnosis of Ehlers-Danlos Syndrome
Diagnosing Ehlers-Danlos Syndrome (EDS) involves a combination of clinical assessment, genetic testing, and family history analysis. Since there are several types of EDS, each with different genetic mutations, it is essential to accurately identify the specific subtype in order to provide the best management strategy.
Clinical Assessment
The diagnosis of EDS often begins with a detailed clinical evaluation, which includes a review of the patient’s symptoms and family medical history. Key diagnostic features include hyperelastic skin, joint hypermobility, and a history of frequent dislocations or joint instability. A physical examination is conducted to assess skin texture, joint flexibility, and the presence of characteristic bruising or scarring. The Beighton score, a system used to measure joint hypermobility, may be used as part of this assessment.
Genetic Testing
Genetic testing plays a crucial role in confirming the diagnosis and identifying the specific mutation responsible for the disorder. Whole-exome sequencing or targeted genetic tests are used to identify mutations in genes related to collagen synthesis and processing, such as COL1A1, COL1A2, COL3A1, and others, depending on the type of EDS suspected. These tests can help differentiate between the various subtypes of EDS and rule out other connective tissue disorders.
Family History and Genetic Counseling
Because EDS is inherited in an autosomal dominant or recessive pattern, understanding the family history is essential. If there is a history of EDS or related connective tissue disorders, it strengthens the suspicion of the condition. Genetic counseling may be offered to the family, especially when a specific mutation is identified, to discuss inheritance patterns, recurrence risks, and prenatal testing options for future pregnancies.
Skin Biopsy and Imaging
In some cases, a skin biopsy or imaging studies may be performed to assess the integrity of connective tissues or to rule out other causes of similar symptoms, such as other collagenopathies or connective tissue disorders.
Treatment Regimens for Ehlers-Danlos Syndrome
There is no cure for Ehlers-Danlos Syndrome (EDS), so treatment focuses on managing symptoms, preventing complications, and improving quality of life. The treatment regimen varies based on the subtype of EDS and the specific symptoms the patient experiences.
Multidisciplinary Approach
A multidisciplinary team of healthcare providers, including geneticists, dermatologists, orthopedic specialists, and physical therapists, is often involved in managing EDS. This team approach ensures that all aspects of the condition are addressed, including skin care, joint stability, and cardiovascular health.
Joint and Skin Care
Due to joint hypermobility, individuals with EDS are at higher risk for joint dislocations and sprains. Physical therapy plays a critical role in strengthening muscles around the joints to reduce the risk of injury. Bracing or splinting may be used to provide additional support and stability to hypermobile joints. Patients are also advised to avoid high-impact activities that could lead to joint trauma. Skin care is also important, as patients may experience hyperelasticity and fragile skin prone to bruising and scarring. Gentle skin care routines and protective measures, such as avoiding excessive friction and trauma, can help minimize skin injuries.
Pain Management
Chronic pain is a common symptom of EDS, especially in patients with frequent joint dislocations and soft tissue damage. Non-steroidal anti-inflammatory drugs (NSAIDs), acetaminophen, and other analgesics may be used to manage pain. In some cases, patients may benefit from corticosteroid injections or other pain-relieving treatments.
Cardiovascular Management
For patients with vascular EDS, regular monitoring of cardiovascular health is essential. Blood pressure management and surveillance for arterial abnormalities, such as aneurysms, are critical components of treatment. Lifestyle modifications, such as avoiding smoking and maintaining a healthy weight, are encouraged to reduce cardiovascular risk.
Surgical Interventions
In some cases, surgical procedures may be necessary to repair joint or vascular damage. However, surgery should be approached with caution due to the increased risk of complications such as wound healing issues and bleeding. Careful planning and coordination with the healthcare team are essential to minimize risks.
Complications of Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome (EDS) can lead to a variety of complications, some of which are potentially life-threatening. These complications arise due to the connective tissue abnormalities that affect various body systems, including the skin, joints, and blood vessels. The severity and range of complications depend on the specific subtype of EDS.
Joint and Musculoskeletal Issues
The most common complications of EDS involve the musculoskeletal system, especially due to joint hypermobility and skin hyperelasticity. Individuals with EDS are prone to frequent joint dislocations, sprains, and fractures. Chronic pain from musculoskeletal problems is another significant issue, as damaged tissues and joints can lead to long-term discomfort. In some cases, osteoarthritis may develop earlier than usual due to joint instability.
Vascular Complications
For those with vascular EDS, one of the most serious complications is the risk of arterial rupture or aneurysms. The fragile blood vessels are more susceptible to damage, and even minor trauma can lead to life-threatening internal bleeding. These individuals may also experience spontaneous rupture of internal organs, such as the intestines or uterus, which can be fatal without prompt medical attention.
Skin Complications
Individuals with EDS often have highly fragile skin, which can lead to frequent bruising, scarring, and delayed wound healing. Due to the skin’s hyperelasticity, wounds may stretch or tear easily, and even minor cuts or abrasions can result in significant scarring. In severe cases, skin fragility can lead to spontaneous skin rupture, requiring careful wound management.
Cardiovascular and Internal Organ Damage
In addition to vascular complications, individuals with EDS may experience internal organ damage due to the fragility of connective tissues. This can lead to complications such as lung or gastrointestinal perforations, which require immediate medical intervention to prevent life-threatening consequences.
Neurological Issues
EDS can also affect the nervous system. Individuals may experience chronic headaches, nerve compression, or even issues with proprioception, where the body loses the ability to sense its position in space. These neurological complications can significantly impact daily functioning and overall quality of life.
Prevention of Complications
Patient Education
Patient education plays a vital role in managing Ehlers-Danlos Syndrome (EDS) and improving the quality of life for individuals affected by the condition. As EDS is a lifelong disorder, educating patients and their families about the nature of the disease, its symptoms, and management strategies is essential for both physical and emotional well-being.
Understanding EDS and Its Types
Patients should be educated about the specific type of EDS they have, as there are various subtypes, each with different manifestations and risks. Understanding the nature of the disease, including the genetic causes and inheritance patterns, helps individuals manage expectations and make informed decisions about family planning. This knowledge also empowers patients to take an active role in managing their health.
Symptom Awareness and Early Intervention
Patients should be encouraged to recognize common symptoms such as joint hypermobility, skin fragility, and easy bruising. Early detection of complications, such as joint dislocations, cardiovascular issues, or skin injuries, can reduce the risk of severe outcomes. Patients should be aware of warning signs and seek medical attention promptly if complications arise, particularly those related to the heart and blood vessels.
Joint Protection and Physical Therapy
Education on joint protection is crucial, especially for individuals with the hypermobility type of EDS. Patients should learn safe techniques for daily activities to avoid joint dislocations or strains. Physical therapy, including strengthening exercises, can help stabilize joints and improve muscle tone, reducing the likelihood of musculoskeletal injuries. Occupational therapy can also teach adaptive strategies for managing activities of daily living.
Skin Care and Wound Management
Because individuals with EDS have fragile skin, educating patients on proper skin care is important. This includes moisturizing regularly, using mild soaps, and protecting the skin from sun damage. Instructing patients on effective wound care techniques, such as cleaning and bandaging minor cuts, can help prevent infections and reduce scarring.
Psychosocial Support and Coping Strategies
EDS can be physically and emotionally challenging, so providing patients with information on coping strategies is essential. Encouraging them to seek psychological support or counseling may help them manage the mental strain of living with a chronic condition. Support groups and online communities can also offer a valuable source of emotional support, as sharing experiences with others facing similar challenges can reduce feelings of isolation.
By providing comprehensive education about EDS, healthcare professionals can empower patients to manage the condition proactively and improve their overall quality of life.
Prognosis
The prognosis of Ehlers-Danlos Syndrome (EDS) largely depends on the specific type of EDS a patient has, as well as the severity of symptoms and any associated complications. Since EDS is a genetic disorder affecting connective tissues, it is a lifelong condition, but the course of the disease varies greatly between individuals.
Impact of EDS Type on Prognosis
The different subtypes of EDS influence prognosis in distinct ways. For example, individuals with the hypermobility type (hEDS) often have a relatively normal life expectancy and can lead independent lives with proper management, though they may experience chronic pain, joint instability, and frequent dislocations. In contrast, individuals with vascular EDS (vEDS), caused by mutations affecting blood vessels, may have a significantly reduced life expectancy due to the risk of arterial rupture and internal bleeding. Early diagnosis and management can improve survival in these cases, but the risks associated with vascular complications remain high.
Management of Symptoms and Complications
The prognosis for patients with EDS can improve with early diagnosis, proper symptom management, and a tailored care plan. Regular monitoring for potential complications, such as heart issues or musculoskeletal problems, can prevent severe outcomes. With physical therapy, joint protection strategies, and proper wound care, many individuals with milder forms of EDS can live relatively normal lives. However, severe forms of the condition may require more intensive care, including surgical intervention and ongoing support from multidisciplinary teams.
Quality of Life Considerations
Although life expectancy may not be significantly impacted in most cases, the quality of life can be affected by chronic pain, mobility challenges, and the emotional toll of dealing with a lifelong condition. Ongoing psychological support, community engagement, and proper education on disease management can greatly improve the well-being of patients and enhance their coping mechanisms.
Conclusion
Ehlers-Danlos syndrome is a complex group of connective tissue disorders characterized by hypermobility, skin hyper-elasticity, and increased susceptibility to bruising and injury. The causes of this condition are often genetic, with various mutations in collagen and other connective tissue-related genes leading to different forms of the syndrome. Its symptoms can range from mild to severe, affecting the skin, joints, and blood vessels. Pathophysiologically, the impaired collagen production and structure contribute to tissue fragility, leading to the common clinical manifestations. Diagnosis relies heavily on clinical evaluation, family history, and genetic testing, while management focuses on symptom relief and preventing complications such as joint dislocations or vascular issues. Although no cure exists, early intervention and appropriate treatment can improve quality of life and reduce the risk of severe complications. Patient education and regular monitoring are crucial for managing the condition effectively, ensuring patients lead active, healthy lives.
References
Mayo Clinic. (2023). Ehlers-Danlos syndrome. Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/ehlers-danlos-syndrome
National Library of Medicine. (2023). Ehlers-Danlos syndrome overview. National Institutes of Health. https://www.ncbi.nlm.nih.gov
Genetics Home Reference. (2023). Ehlers-Danlos syndromes. U.S. National Library of Medicine. https://ghr.nlm.nih.gov
Ehlers-Danlos Society. (2023). What is EDS? The Ehlers-Danlos Society. https://www.ehlers-danlos.com