Nursing Paper Example on Epidermolysis Bullosa
Nursing Paper Example on Epidermolysis Bullosa
Epidermolysis bullosa (EB) is a rare group of inherited skin disorders characterized by blister formation and skin fragility. The condition arises from mutations in genes responsible for the proteins that bind skin layers together. This leads to skin that is easily damaged by friction or trauma, causing blisters and sores. The severity of EB can vary significantly, with some forms presenting mild skin fragility and others causing widespread blistering, scarring, and systemic complications. EB can affect not only the skin but also mucous membranes, including those of the mouth, eyes, and gastrointestinal tract. Individuals with EB face challenges that extend beyond the physical symptoms, as the condition can impact quality of life and require ongoing care to manage wounds and prevent complications.
Causes
Epidermolysis bullosa is primarily caused by genetic mutations in genes that produce structural proteins involved in the attachment of skin layers. These include mutations in the COL7A1 gene responsible for type VII collagen, which plays a critical role in anchoring the skin’s layers. Inherited as either an autosomal dominant or recessive trait, the different forms of EB are classified based on the type of gene mutation and the layer of skin affected. In simplex EB, the mutations primarily affect the basal cells of the epidermis, while dystrophic EB involves mutations in type VII collagen. Junctional EB results from mutations in genes encoding proteins that form the basement membrane zone. There are also rare forms, such as Kindler syndrome, caused by mutations in the FERMT1 gene.
The severity of the condition correlates with the nature of the mutation. Autosomal dominant inheritance typically results in a less severe form, while autosomal recessive inheritance often leads to more severe manifestations, such as extensive blistering and scarring.
Signs and Symptoms
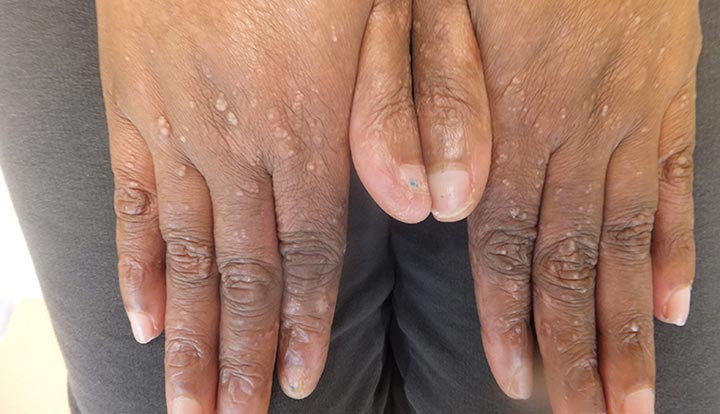
The primary feature of epidermolysis bullosa is the formation of blisters after even minimal trauma. The severity and location of blisters depend on the type of EB. In epidermolysis bullosa simplex, blisters often form on the hands, feet, and knees, with minimal scarring. Dystrophic EB is characterized by deeper blisters, which heal with scarring, leading to contractures and deformities. In junctional EB, the blisters may appear in the mouth and other mucosal surfaces, with a tendency for severe systemic complications. Blistering can occur spontaneously or as a result of friction, such as rubbing or pressure. The affected skin appears fragile, and wounds may heal slowly or with increased scarring. In more severe cases, complications such as infections, skin cancer, and difficulty with swallowing or breathing can occur.
The chronic nature of the disease leads to ongoing issues with wound care, pain management, and infections. In some forms of EB, such as dystrophic EB, patients may develop contractures, deformities, and shortened limbs due to extensive scarring. These physical symptoms are often compounded by psychological impacts, including social and emotional stress, due to the visible nature of the condition and the need for continuous medical attention.
Etiology
The etiology of epidermolysis bullosa is rooted in genetic mutations that disrupt the skin’s ability to maintain its integrity. These mutations typically affect structural proteins that are involved in maintaining the cohesion of the epidermis and dermis. In epidermolysis bullosa simplex, mutations often involve keratins, proteins that provide strength and resilience to the skin. In dystrophic EB, the genetic mutations affect type VII collagen, a protein that forms anchoring fibrils that secure the epidermis to the dermis. This instability leads to mechanical fragility in the skin.
The onset and severity of symptoms depend on the specific gene mutation and its inheritance pattern. In junctional EB, mutations in laminin or collagen XVII lead to defects in the basement membrane, disrupting the connection between the epidermis and dermis. In Kindler syndrome, a form that involves both blistering and photosensitivity, mutations in the FERMT1 gene impair the skin’s response to UV light, contributing to both mechanical fragility and photosensitivity.
Pathophysiology
The pathophysiology of epidermolysis bullosa involves a breakdown in the structural integrity of the skin layers due to defective adhesion molecules or structural proteins. In the case of epidermolysis bullosa simplex, defects in keratins, which provide mechanical strength to skin cells, make the epidermis prone to mechanical injury. The loss of keratin leads to the formation of fluid-filled blisters, which rupture easily.
In dystrophic EB, mutations in type VII collagen disrupt the structural framework between the epidermis and dermis. This weakens the anchoring fibrils that hold these layers together, making the skin prone to separation. When trauma occurs, the layers of skin pull apart, forming large blisters that often lead to scarring. Junctional EB results from defects in the proteins of the basement membrane zone, including laminins and collagen XVII, which form the interface between the epidermis and dermis. These defects contribute to blistering, not just at the skin surface but also within mucosal areas such as the mouth and eyes.
The severity of the disease depends on whether the mutations cause partial or complete loss of function in the affected proteins. In Kindler syndrome, defective FERMT1 impairs cell-cell and cell-matrix interactions, leading to increased skin fragility and photosensitivity.
DSM-5 Diagnosis
While there is no specific diagnostic criterion for epidermolysis bullosa in the DSM-5, diagnosis typically relies on clinical presentation and genetic testing. A skin biopsy may be performed to determine the type of EB by examining the ultrastructure of the skin and identifying the presence of blisters in the appropriate skin layers. Immunofluorescence studies can also identify specific proteins affected by mutations. Genetic testing is the most definitive method for diagnosing EB, allowing for identification of the specific gene mutation and confirming the type of EB.
Treatment Regimens
The primary approach to treating epidermolysis bullosa involves symptom management and preventing complications. Because EB is a genetic disorder, there is currently no cure, and treatment focuses on wound care, pain relief, and infection prevention. Regular wound care, including the use of non-stick bandages and moisturizers, is essential for managing blisters. Infection control is a critical aspect of care, as open wounds are susceptible to bacterial infections. Topical antibiotics and oral antibiotics may be used to treat infections.
In more severe cases, surgical intervention may be necessary to release contractures or repair deformities caused by scarring. Stem cell therapy has shown promise in experimental treatments, where stem cells may help regenerate healthy skin. Genetic therapies are being researched as potential future treatments, though they are not yet widely available.
Complications
Complications from epidermolysis bullosa can be severe, depending on the type and severity of the condition. Chronic wounds can lead to infections, which may spread to deeper tissues or even the bloodstream, causing sepsis. In dystrophic EB, scarring can result in contractures, deformities, and loss of function in affected limbs. Individuals with EB are also at higher risk of developing skin cancer, particularly squamous cell carcinoma, due to chronic UV exposure and ongoing skin damage.
In cases of junctional EB, the involvement of mucosal surfaces can lead to serious complications, such as difficulty swallowing, respiratory problems, and corneal scarring, which can cause vision loss. Chronic anemia, malnutrition, and growth retardation may also result from the difficulty in maintaining proper nutrition due to oral and esophageal blistering.
Prevention
Preventing epidermolysis bullosa is not possible because it is a genetic condition. However, individuals with a family history of EB may benefit from genetic counseling to understand the risks of passing the condition to offspring. Early diagnosis through genetic testing can help families understand the specific type of EB and the severity of the disease, which may inform treatment options.
While the condition itself cannot be prevented, preventing complications is a crucial aspect of management. Protective measures, such as avoiding trauma to the skin and using gentle skin care products, can help minimize blister formation. Regular follow-up with healthcare providers is essential to manage wounds, infections, and other complications.
Patient Education
Education is a key component of managing epidermolysis bullosa. Patients and caregivers should be taught proper wound care techniques, including cleaning and dressing blisters to prevent infection. Using soft fabrics, wearing padded shoes, and avoiding friction are critical for reducing skin trauma. Patients should be informed about the importance of protecting their skin from sun exposure to reduce the risk of skin cancer.
Family members should be educated on the psychological impacts of living with a chronic, visible condition. Support groups and counseling may provide emotional support and coping strategies for both patients and caregivers. Nutrition management is also essential, particularly in cases where oral involvement makes eating difficult.
Conclusion
Epidermolysis bullosa is a group of inherited disorders that cause skin fragility and blistering, leading to chronic pain, infections, and potential deformities. While the condition cannot be cured, early diagnosis, careful management of symptoms, and prevention of complications can improve the quality of life for individuals affected by EB. Continued research into genetic therapies and advanced wound care techniques holds promise for the future treatment of this challenging condition.
References
Epidermolysis Bullosa Research Partnership. (2023). Epidermolysis bullosa overview. https://www.ebresearch.org/overview
Mayo Clinic. (2023). Epidermolysis bullosa. https://www.mayoclinic.org/diseases-conditions/epidermolysis-bullosa
National Institute of Arthritis and Musculoskeletal and Skin Diseases. (2023). Epidermolysis bullosa. https://www.niams.nih.gov/health-topics/epidermolysis-bullosa
The Ehlers-Danlos Society. (2023). Epidermolysis bullosa. https://www.ehlers-danlos.com/epidermolysis-bullosa
Do you need a similar assignment done for you from scratch? Order now!
Use Discount Code "Newclient" for a 15% Discount!