Nursing Paper Example on Gaucher Disease
Nursing Paper Example on Gaucher Disease
Gaucher disease is a rare inherited lysosomal storage disorder caused by a deficiency in the enzyme beta-glucocerebrosidase. This enzyme’s dysfunction leads to the accumulation of a fatty substance called glucocerebroside in various cells and tissues, causing multi-organ dysfunction. The disease is most prevalent among Ashkenazi Jews, although it occurs worldwide. There are three primary subtypes of Gaucher disease, classified based on the presence and progression of neurological symptoms. Advances in therapeutic approaches have significantly enhanced disease management and patient outcomes, although challenges remain for more severe forms.
Causes
The underlying cause of Gaucher disease is mutations in the GBA gene, which provides instructions for producing beta-glucocerebrosidase. This enzyme is essential for breaking down glucocerebroside into glucose and ceramide within lysosomes. When the enzyme is deficient or defective, glucocerebroside accumulates, particularly in macrophages, leading to the formation of Gaucher cells. These cells infiltrate organs such as the liver, spleen, and bone marrow, disrupting their function. The disease follows an autosomal recessive inheritance pattern, meaning two defective copies of the GBA gene—one from each parent—are required for the disease to manifest.
Signs and Symptoms
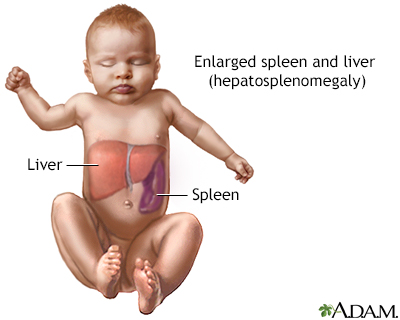
The symptoms of Gaucher disease vary by subtype. Type 1, the most common and non-neurological form, presents with hepatosplenomegaly, anemia, thrombocytopenia, bone pain, and fractures. Fatigue, growth delays, and a predisposition to infections are also observed. In type 2, the acute neuropathic form, symptoms manifest early in infancy and include severe neurological decline, spasticity, seizures, and difficulty swallowing. This form is typically fatal by age two. Type 3, the chronic neuropathic form, presents with systemic symptoms similar to type 1 but with progressive neurological impairment, such as ataxia, cognitive decline, and ocular abnormalities. Complications like pulmonary hypertension and an increased risk of malignancies, particularly multiple myeloma, are common across all forms.
Etiology
The etiology of Gaucher disease is directly linked to the biochemical and genetic defects caused by mutations in the GBA gene. These mutations hinder the normal degradation of glucocerebroside, leading to its pathological accumulation. The resultant Gaucher cells contribute to inflammation, organomegaly, and bone disease. Environmental and genetic factors, such as the specific type of mutation, influence the severity and type of Gaucher disease. Certain mutations, such as N370S, are associated with milder forms, whereas others like L444P are linked to severe neurological involvement.
Pathophysiology
The pathological hallmark of Gaucher disease is the presence of Gaucher cells, which are engorged macrophages containing undigested glucocerebroside. These cells infiltrate organs like the spleen, liver, bone marrow, and, in neuropathic types, the central nervous system. Their presence triggers inflammation and disrupts normal tissue architecture. In the bones, Gaucher cells interfere with vascularization and remodeling, leading to pain and fractures. In the nervous system, accumulated substrates disrupt lysosomal function, causing neuronal death and progressive neurodegeneration. The systemic inflammatory state induced by Gaucher cells further exacerbates organ dysfunction and contributes to disease complications.
Diagnosis
Gaucher disease diagnosis begins with clinical suspicion based on symptoms such as unexplained splenomegaly or bone pain. Enzyme assay testing is the gold standard, measuring beta-glucocerebrosidase activity in leukocytes or fibroblasts. Genetic testing identifies GBA mutations, confirming the diagnosis and assisting in subtype classification. Imaging studies, such as magnetic resonance imaging (MRI) or ultrasound, help evaluate organ involvement and monitor disease progression. Biomarkers like chitotriosidase and glucosylsphingosine levels are elevated in Gaucher disease and are valuable for tracking treatment response and disease activity.
Treatment Regimens
The management of Gaucher disease depends on its type and severity. Enzyme replacement therapy (ERT) is the cornerstone treatment for type 1 and some aspects of type 3 disease. Intravenous administration of recombinant beta-glucocerebrosidase, such as imiglucerase or velaglucerase, effectively reduces glucocerebroside accumulation, alleviating systemic symptoms. Substrate reduction therapy (SRT) with oral agents like eliglustat offers an alternative, especially for patients who cannot tolerate ERT. For neurological symptoms in type 3, interventions are largely supportive, as ERT cannot cross the blood-brain barrier. Palliative care is the mainstay for type 2, focusing on symptom management and quality of life. Additional therapies include blood transfusions for anemia, bisphosphonates for bone health, and pain management. Hematopoietic stem cell transplantation (HSCT) is considered in severe cases but carries significant risks.
Patient Education
Patient education plays a vital role in managing Gaucher disease. Patients and families must understand the genetic basis of the disease and the implications for family planning, including the importance of genetic counseling. Adherence to treatment regimens, whether ERT or SRT, is critical for symptom control and preventing complications. Patients should recognize early signs of disease progression, such as worsening fatigue or bone pain, to seek timely medical intervention. Lifestyle modifications, such as maintaining a balanced diet, regular exercise, and avoiding smoking, can help improve overall health and reduce disease complications.
Research and Future Directions
Ongoing research in Gaucher disease aims to address limitations in current therapies and uncover new treatment strategies. Gene therapy is being explored to provide a long-term cure by correcting the underlying genetic defect. Pharmacological chaperones, which stabilize misfolded beta-glucocerebrosidase, show promise in restoring enzyme function. Investigations into biomarkers and imaging techniques aim to improve disease monitoring and individualized treatment plans. Understanding the association between GBA mutations and Parkinson’s disease may provide insights into shared pathophysiological mechanisms, potentially benefiting both conditions.
Conclusion
Gaucher disease is a complex and multisystemic lysosomal storage disorder that poses significant challenges for patients and healthcare providers. While advancements in enzyme replacement and substrate reduction therapies have improved outcomes, severe neurological forms of the disease remain difficult to manage. Early diagnosis, personalized treatment strategies, and ongoing research are essential to optimize care for individuals with Gaucher disease. Continued education and multidisciplinary support are key to enhancing the quality of life for affected patients and their families.
References
Cox, T. M. (2023). Gaucher disease: Clinical features and current treatment. The Lancet. https://www.thelancet.com
Zimran, A., & Elstein, D. (2023). Enzyme replacement and substrate reduction in Gaucher disease. Orphanet Journal of Rare Diseases. https://ojrd.biomedcentral.com
Balwani, M., & Desnick, R. J. (2023). Genetic basis of Gaucher disease. Genetics in Medicine. https://geneticsinmedicine.org
Grabowski, G. A., & Mistry, P. K. (2023). Pathophysiology of Gaucher disease. Nature Reviews Endocrinology. https://nature.com/nrendocrin
de Fost, M., & Aerts, J. M. (2023). Advances in Gaucher biomarkers and therapies. Journal of Inherited Metabolic Disease. https://onlinelibrary.wiley.com
Do you need a similar assignment done for you from scratch? Order now!
Use Discount Code "Newclient" for a 15% Discount!