Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder
Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder

Causes
Huntington’s disease (HD) is primarily caused by a mutation in the HTT gene located on chromosome 4. This mutation involves an abnormal repetition of the CAG trinucleotide sequence within the gene, leading to the production of an altered form of the huntingtin protein. Normally, this protein plays a crucial role in neuronal function and survival, but the mutated version results in its abnormal accumulation within neurons.
The inheritance pattern of HD follows an autosomal dominant pattern, meaning that a person needs only one copy of the mutated gene from either parent to develop the disease. As a result, each child of an affected parent has a 50% chance of inheriting the mutated gene and eventually developing HD.
The number of CAG repeats in the HTT gene correlates with the age of onset and severity of HD symptoms. Individuals with fewer repeats tend to develop symptoms later in life and have a milder form of the disease, while those with a higher number of repeats typically experience an earlier onset and more severe symptoms.
Although the exact mechanism by which the mutated huntingtin protein leads to neuronal dysfunction and death is not fully understood, research suggests that it disrupts various cellular processes within neurons. This disruption includes impaired protein degradation, mitochondrial dysfunction, excitotoxicity, and altered neurotransmitter signaling.
Furthermore, studies indicate that the mutated huntingtin protein may interfere with the function of other proteins and cellular pathways involved in neuronal survival, ultimately contributing to the progressive neurodegeneration observed in HD.
Overall, the underlying cause of Huntington’s disease lies in the abnormal expansion of the CAG repeats within the HTT gene, resulting in the production of a mutated huntingtin protein and subsequent neuronal dysfunction and death. (Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder)
Signs and Symptoms
Huntington’s disease (HD) manifests with a wide array of symptoms that affect various aspects of an individual’s physical and cognitive functioning. These symptoms typically begin to appear between the ages of 30 and 50, although onset can occur at any age, including childhood or late adulthood.
:max_bytes(150000):strip_icc()/huntingtons-disease-symptoms-5091956-Final-c6e5d478c42945b593bafa65d9408e23.jpg)
One of the hallmark features of HD is involuntary movements, known as chorea. These movements are characterized by jerky, random, and uncontrollable motions that affect the limbs, face, and trunk. Chorea tends to worsen as the disease progresses, leading to difficulties in coordination and balance.
Cognitive impairment is another prominent feature of HD, affecting memory, judgment, and executive function. Individuals may experience difficulties with planning, organizing, and completing tasks, as well as a decline in overall cognitive abilities. Additionally, changes in personality and behavior are common, with individuals exhibiting impulsivity, irritability, and mood swings. Psychiatric symptoms such as depression and anxiety are also prevalent in individuals with HD.
Motor symptoms beyond chorea include rigidity, dystonia, and bradykinesia, which can further impair mobility and coordination. As the disease advances, individuals may experience difficulties with speech and swallowing, leading to aspiration pneumonia and other complications.
Furthermore, HD often leads to significant functional decline, impacting activities of daily living and reducing independence. Individuals may require assistance with basic tasks such as dressing, eating, and personal hygiene.
Overall, the signs and symptoms of Huntington’s disease encompass a broad spectrum of motor, cognitive, and psychiatric impairments that progressively worsen over time. Early recognition and management of these symptoms are crucial in providing optimal care and support for individuals living with HD. (Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder)
Etiology
The etiology of Huntington’s disease (HD) is rooted in a genetic mutation that affects the HTT gene, located on chromosome 4. This mutation involves an abnormal expansion of the CAG trinucleotide repeat sequence within the gene, resulting in the production of an altered form of the huntingtin protein.
The normal function of the huntingtin protein is essential for neuronal health and survival, but the mutated version leads to detrimental consequences within neurons. Specifically, the abnormal huntingtin protein undergoes misfolding and aggregation, forming insoluble clumps within neurons.
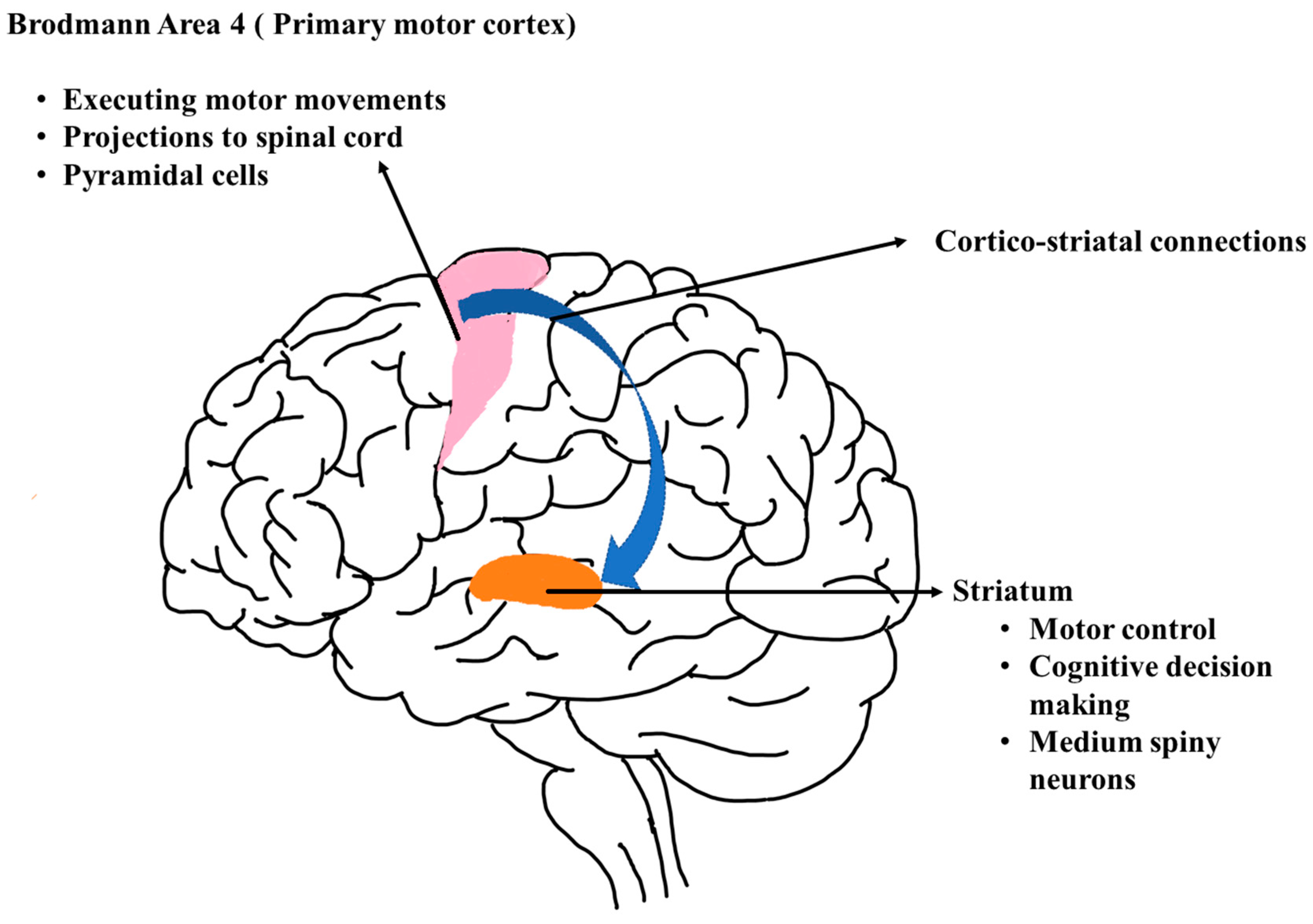
These protein aggregates disrupt various cellular processes, including protein degradation pathways, mitochondrial function, and intracellular signaling pathways. This disruption ultimately leads to impaired neuronal function and eventual cell death, particularly in regions of the brain involved in movement control, cognition, and behavior.
Furthermore, research suggests that the mutated huntingtin protein may exert toxic effects on neighboring neurons through mechanisms such as excitotoxicity, oxidative stress, and inflammation. These secondary processes contribute to the progressive neurodegeneration observed in HD.
The exact mechanisms by which the mutated huntingtin protein leads to neuronal dysfunction and death are complex and multifaceted. Studies continue to explore the intricate molecular pathways involved in HD pathogenesis, with the goal of identifying potential targets for therapeutic intervention.
Additionally, while the presence of the mutated HTT gene is necessary for the development of HD, other genetic and environmental factors may modulate disease onset and progression. Factors such as genetic modifiers, epigenetic changes, and lifestyle factors may influence the age of symptom onset and the severity of symptoms in individuals with HD.
Overall, the etiology of Huntington’s disease encompasses a complex interplay of genetic, molecular, and environmental factors that contribute to the pathogenesis of this devastating neurodegenerative disorder. (Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder)
Pathophysiology
The pathophysiology of Huntington’s disease (HD) revolves around the progressive dysfunction and degeneration of neurons within specific regions of the brain, particularly the basal ganglia and cerebral cortex. This neurodegenerative process is driven by the accumulation of mutant huntingtin protein, resulting from the abnormal expansion of the CAG trinucleotide repeat sequence within the HTT gene.
The mutated huntingtin protein undergoes misfolding and aggregation, forming insoluble clumps within neurons. These protein aggregates disrupt various cellular processes, including protein degradation pathways, mitochondrial function, and intracellular signaling pathways.
One key aspect of HD pathophysiology is the disruption of neurotransmitter signaling within neuronal circuits. The basal ganglia, which are critical for motor control, exhibit altered dopamine neurotransmission due to dysfunction in the cortico-striatal pathway. This imbalance in neurotransmitter signaling contributes to the characteristic motor symptoms of HD, including chorea and dystonia.
Moreover, the cerebral cortex, responsible for higher cognitive functions, is also affected by HD pathology. Neuronal loss and atrophy in cortical regions lead to cognitive decline and psychiatric symptoms, including impairments in memory, executive function, and emotional regulation.
In addition to neuronal dysfunction, HD pathophysiology involves widespread cellular damage and neuroinflammation. Reactive gliosis, microglial activation, and cytokine release contribute to a neurotoxic environment, further exacerbating neuronal injury and degeneration.
Furthermore, emerging evidence suggests that non-cell autonomous mechanisms, involving interactions between neurons, glia, and other cell types, play a crucial role in HD pathophysiology. These interactions contribute to the spreading of pathology throughout the brain and the amplification of neurodegenerative processes.
Overall, the pathophysiology of Huntington’s disease encompasses a complex interplay of molecular, cellular, and circuit-level dysfunctions, ultimately leading to progressive neurodegeneration and the clinical manifestations of the disease. Understanding these underlying mechanisms is essential for the development of targeted therapeutic strategies aimed at slowing or halting disease progression. (Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder)
DSM-5 Diagnosis
The diagnosis of Huntington’s disease (HD) follows specific criteria outlined in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5). These criteria help clinicians identify individuals with HD based on their clinical presentation and genetic testing results.
- Motor Abnormalities: The DSM-5 criteria for HD require the presence of motor abnormalities consistent with the disease. These may include chorea, dystonia, or other involuntary movements that interfere with daily functioning.
- Cognitive Decline: HD often involves cognitive impairment, which may manifest as difficulties with memory, executive function, and other cognitive domains. Clinically significant cognitive decline must be present to meet the diagnostic criteria.
- Psychiatric Symptoms: Individuals with HD commonly experience psychiatric symptoms such as depression, anxiety, irritability, and impulsivity. These symptoms contribute to the overall clinical picture and are considered in the diagnostic evaluation.
- Family History: A family history of HD is a crucial component of the diagnostic process. Since HD is an autosomal dominant disorder, a positive family history increases the likelihood of the diagnosis.
- Genetic Testing: Confirmatory genetic testing is typically performed to establish the presence of the mutated HTT gene. A positive genetic test result in combination with the clinical symptoms is necessary to confirm the diagnosis of HD.
It is important to note that the diagnosis of HD requires careful clinical assessment by a qualified healthcare professional, including a detailed medical history, neurological examination, and consideration of relevant laboratory and imaging studies. Additionally, genetic counseling and testing play a significant role in confirming the diagnosis and providing information about the inheritance pattern and implications for family members. (Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder)
Treatment Regimens and Patient Education:
While there is currently no cure for Huntington’s disease (HD), treatment focuses on managing symptoms, improving quality of life, and providing comprehensive care and support for patients and their families. A multidisciplinary approach involving healthcare professionals from various specialties is essential in addressing the complex needs of individuals with HD.
1. Medication Management:
- Medications such as tetrabenazine and deutetrabenazine are commonly prescribed to help control chorea and other involuntary movements associated with HD.
- Antipsychotic medications may be used to manage psychiatric symptoms such as depression, anxiety, and psychosis.
- Antidepressants and mood stabilizers may also be prescribed to address mood disturbances and behavioral symptoms.
2. Physical and Occupational Therapy:
- Physical therapy can help maintain mobility, improve balance, and manage motor symptoms.
- Occupational therapy focuses on adapting the environment and developing strategies to maximize independence in activities of daily living.
3. Speech and Swallowing Therapy:
- Speech therapy may be beneficial for individuals experiencing difficulties with speech and swallowing.
- Speech therapists can provide exercises and techniques to improve communication and swallowing function.
4. Nutritional Support:
- As swallowing difficulties can lead to malnutrition and dehydration, nutritional counseling and support are essential in managing these issues.
- Speech therapists and dietitians can work together to develop strategies to ensure adequate nutrition and hydration.
5. Psychological Support and Counseling:
- Psychologists or counselors can provide individual and family counseling to address emotional and psychological challenges associated with HD.
- Support groups and peer support networks can offer valuable emotional support and practical advice for individuals and families coping with HD.
Patient Education:
- Educating patients and their families about the progressive nature of HD, available treatments, and the importance of regular medical follow-up is crucial.
- Providing information about community resources, support services, and advocacy organizations can help patients and families access additional support and assistance.
- Genetic counseling and testing should be offered to individuals at risk of HD to provide information about the inheritance pattern, genetic testing options, and family planning considerations.
- Encouraging open communication and collaboration between healthcare providers, patients, and families can facilitate shared decision-making and ensure that individual preferences and goals are considered in treatment planning.
A comprehensive approach to the management of Huntington’s disease involves a combination of pharmacological interventions, rehabilitative therapies, nutritional support, psychological counseling, and patient education. By addressing the diverse needs of patients and their families, healthcare professionals can optimize care and support individuals living with HD in maintaining their quality of life and well-being. (Nursing Paper Example on Huntington’s Disease: Understanding a Devastating Neurological Disorder)
Conclusion
Huntington’s disease (HD) presents a significant challenge in the field of neurology, affecting individuals and their families on physical, cognitive, and emotional levels. This essay has explored the causes, signs, etiology, pathophysiology, DSM-5 diagnosis, treatment regimens, and patient education related to HD. Understanding the genetic underpinnings and complex molecular pathways involved in HD pathogenesis is essential for developing targeted therapies aimed at slowing disease progression and improving symptom management. Moreover, a multidisciplinary approach to treatment, encompassing medication management, rehabilitative therapies, nutritional support, psychological counseling, and patient education, is crucial in addressing the diverse needs of individuals with HD. By prioritizing comprehensive care and support, healthcare professionals can empower patients and their families to navigate the challenges of living with HD while maintaining optimal quality of life. Continued research and advancements in the field offer hope for improved outcomes and a better understanding of this debilitating neurodegenerative disorder.
References
https://www.ncbi.nlm.nih.gov/books/NBK559166/
Do you need a similar assignment done for you from scratch? Order now!
Use Discount Code "Newclient" for a 15% Discount!