Nursing Paper Example on Understanding Sickle Cell Disease: An Overview
/in Assignment Help, Assignment Help Nursing, BLOG, Homework Help, Nursing Exam Help, Nursing Paper Help, Solved Nursing Essays /by Aimee GraceNursing Paper Example on Understanding Sickle Cell Disease: An Overview
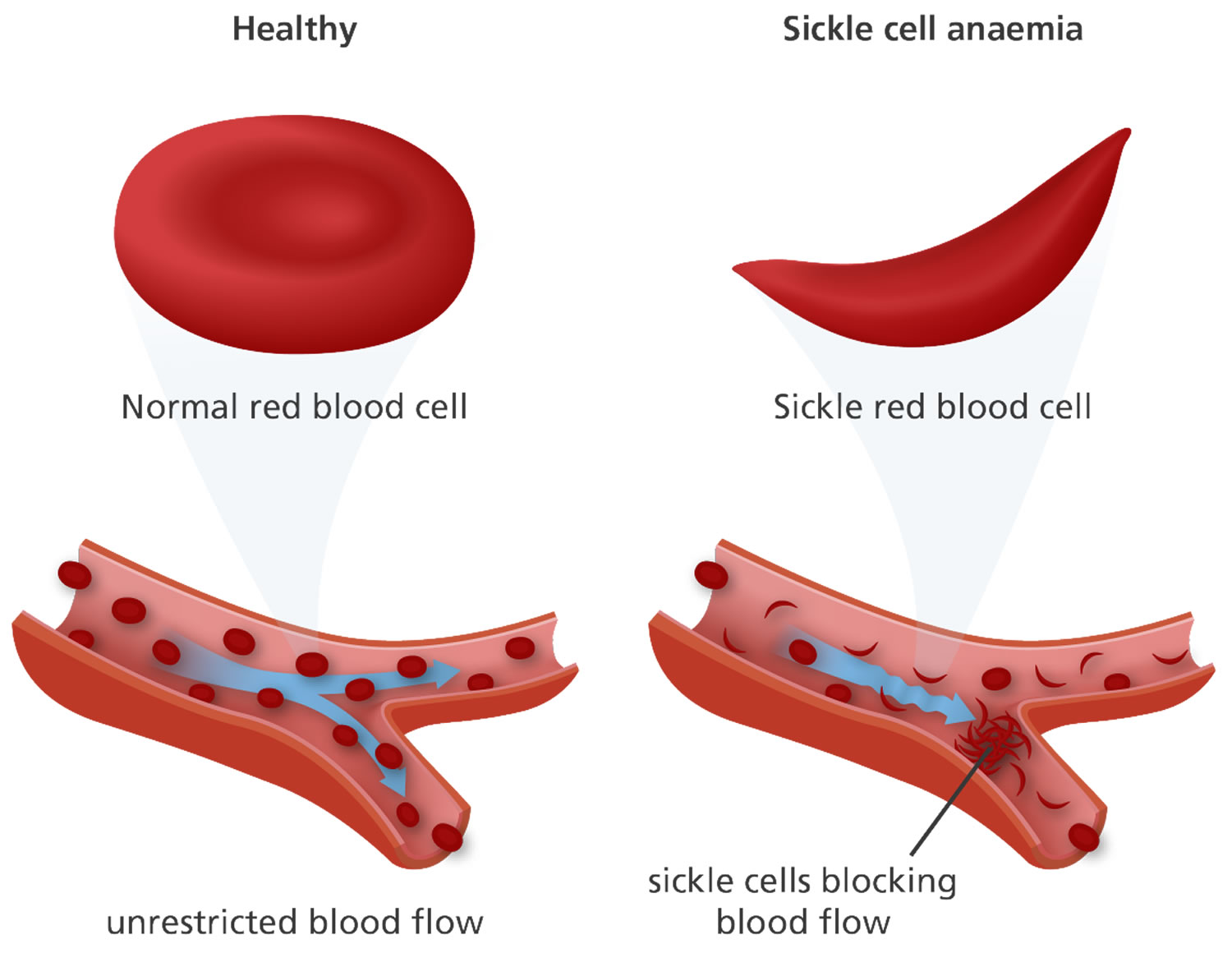
Sickle cell disease (SCD) is a genetic blood disorder that profoundly impacts the lives of millions worldwide. It is particularly prevalent among individuals of African descent but can affect people from diverse ethnic backgrounds. This condition, caused by a mutation in the gene responsible for hemoglobin production, leads to the formation of abnormal hemoglobin known as hemoglobin S (HbS). When oxygen levels are low, HbS causes red blood cells to assume a sickle shape, resulting in a cascade of symptoms and complications. Despite its widespread occurrence, SCD remains a significant public health concern with substantial implications for affected individuals and their families. Understanding the causes, signs, and management of SCD is crucial for effective treatment and improved quality of life for those living with this condition. In this paper, we delve into the various aspects of sickle cell disease, including its causes, symptoms, pathophysiology, diagnosis, treatment, and the importance of patient education. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
Causes of Sickle Cell Disease
Sickle cell disease (SCD) is primarily caused by a genetic mutation affecting the production of hemoglobin, the protein responsible for carrying oxygen in red blood cells. This genetic mutation occurs in the HBB gene, located on chromosome 11, which encodes the beta-globin subunit of hemoglobin. Specifically, SCD is a result of a point mutation in the HBB gene, where thymine is substituted for adenine at the sixth codon, leading to the production of abnormal hemoglobin known as hemoglobin S (HbS).
Individuals inherit SCD through an autosomal recessive inheritance pattern, meaning that both parents must carry a copy of the mutated gene for their child to develop the disease. If both parents are carriers (heterozygous) of the mutated gene, there is a 25% chance with each pregnancy that their child will inherit two copies of the mutated gene, resulting in SCD. If only one parent is a carrier, their child has a 50% chance of inheriting the sickle cell trait (carrying one copy of the mutated gene) and a 50% chance of inheriting a normal hemoglobin gene.
The prevalence of SCD is highest in regions where malaria is endemic, as the sickle cell trait (carrying one copy of the mutated gene) provides some protection against malaria. Consequently, SCD is more common in populations with historical or current exposure to malaria, such as those of African, Mediterranean, Middle Eastern, Indian, and Southeast Asian descent.
Overall, the underlying cause of SCD lies in the genetic mutation affecting hemoglobin production, leading to the formation of abnormal hemoglobin S. Understanding the genetic basis of the disease is crucial for both diagnosis and the development of targeted treatment strategies aimed at managing its symptoms and complications. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
Signs and Symptoms
Sickle cell disease (SCD) manifests through a wide range of signs and symptoms, which can vary in severity and frequency among affected individuals. The most common symptoms of SCD are related to the abnormal behavior of red blood cells containing hemoglobin S (HbS), particularly during episodes of sickling when oxygen levels are low.
One of the hallmark features of SCD is chronic anemia, which results from the decreased lifespan of sickle-shaped red blood cells. Anemia can lead to fatigue, weakness, and pallor, affecting the overall energy levels and quality of life of individuals with SCD.
Pain crises, also known as sickle cell crises, are another characteristic symptom of SCD. These episodes are characterized by sudden and severe pain, often in the bones, joints, abdomen, or chest. Pain crises can be triggered by various factors, including dehydration, infection, stress, and exposure to cold temperatures.
Individuals with SCD are also at increased risk of developing complications such as acute chest syndrome, a potentially life-threatening condition characterized by chest pain, fever, and difficulty breathing. Other complications of SCD include stroke, priapism (prolonged erection), gallstones, and leg ulcers.
Jaundice, caused by the breakdown of red blood cells, is a common finding in individuals with SCD. Jaundice presents as yellowing of the skin and eyes due to elevated levels of bilirubin in the bloodstream.
Moreover, individuals with SCD are more susceptible to infections, particularly those caused by encapsulated bacteria such as Streptococcus pneumoniae. This increased susceptibility to infections is due to functional asplenia (loss of spleen function) resulting from repeated episodes of sickle cell crisis.
Overall, the signs and symptoms of SCD can significantly impact the daily lives and overall health of affected individuals, underscoring the importance of early diagnosis and comprehensive management strategies. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
Etiology of Sickle Cell Disease
Sickle cell disease (SCD) is an inherited genetic disorder caused by a mutation in the HBB gene, which encodes the beta-globin subunit of hemoglobin. This mutation leads to the production of abnormal hemoglobin known as hemoglobin S (HbS), which is responsible for the characteristic sickle shape of red blood cells in individuals with SCD.
The inheritance pattern of SCD follows an autosomal recessive trait, meaning that both parents must carry a copy of the mutated gene for their child to develop the disease. If both parents are carriers (heterozygous) of the mutated gene, there is a 25% chance with each pregnancy that their child will inherit two copies of the mutated gene, resulting in SCD. If only one parent is a carrier, their child has a 50% chance of inheriting the sickle cell trait (carrying one copy of the mutated gene) and a 50% chance of inheriting a normal hemoglobin gene.
The prevalence of SCD varies among different populations, with the highest rates observed in regions where malaria is endemic. Historically, the sickle cell trait (carrying one copy of the mutated gene) has provided a survival advantage against malaria, leading to a higher frequency of the mutated gene in these populations. Consequently, SCD is more common in populations with historical or current exposure to malaria, such as those of African, Mediterranean, Middle Eastern, Indian, and Southeast Asian descent.
While the genetic mutation responsible for SCD has been identified, ongoing research continues to explore the complex interactions between genetic and environmental factors that influence the severity and progression of the disease. Understanding the etiology of SCD is essential for genetic counseling, prenatal screening, and the development of targeted treatment strategies aimed at improving outcomes for individuals affected by this condition. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
Pathophysiology
Sickle cell disease (SCD) is characterized by the abnormal behavior of red blood cells containing hemoglobin S (HbS) under conditions of low oxygen levels. The pathophysiology of SCD is multifaceted and involves several key mechanisms that contribute to the clinical manifestations of the disease.
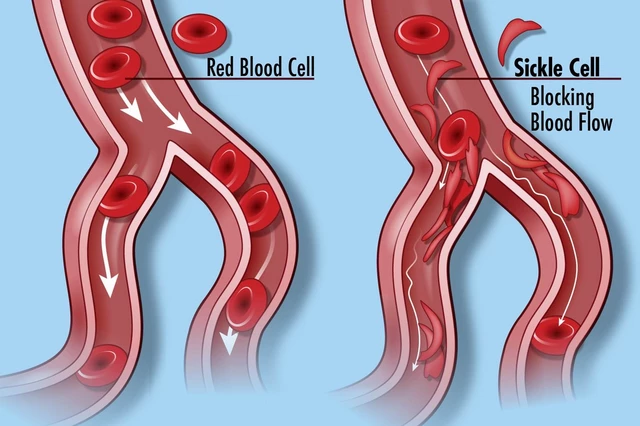
Central to the pathophysiology of SCD is the polymerization of HbS molecules within red blood cells when they are deoxygenated. This polymerization causes the red blood cells to become rigid and assume a sickle shape, impairing their ability to flow smoothly through blood vessels. Consequently, sickled red blood cells can block small blood vessels, leading to tissue ischemia, inflammation, and organ damage.
Moreover, sickled red blood cells have a shortened lifespan compared to normal red blood cells, leading to chronic hemolysis (destruction of red blood cells) and resulting in anemia. The increased destruction of red blood cells releases hemoglobin into the bloodstream, leading to the formation of reactive oxygen species and oxidative stress, further exacerbating tissue damage.
The vaso-occlusive crises, or pain crises, experienced by individuals with SCD result from the obstruction of blood flow by sickled red blood cells, particularly in small blood vessels. These crises can occur spontaneously or be triggered by various factors, including infection, dehydration, stress, or exposure to cold temperatures.
Additionally, the chronic hemolysis associated with SCD leads to the release of free hemoglobin and heme into the bloodstream, which can scavenge nitric oxide, a vasodilator, impairing endothelial function and contributing to vaso-occlusive events and other complications of the disease.
Overall, the pathophysiology of SCD is complex and involves multiple interrelated processes that contribute to the clinical manifestations and complications of the disease. Understanding these underlying mechanisms is essential for the development of targeted therapies aimed at improving outcomes for individuals affected by SCD. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
DMS-5 Diagnosis
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), provides specific criteria for the diagnosis of sickle cell disease (SCD) based on clinical presentation, laboratory findings, and genetic testing.
One of the primary criteria for diagnosing SCD is the presence of sickle-shaped red blood cells on peripheral blood smear examination. Microscopic evaluation of a blood smear allows healthcare providers to visualize the characteristic sickle-shaped morphology of red blood cells, which is indicative of the disease.
Laboratory testing is also crucial for the diagnosis of SCD. Hemoglobin electrophoresis is commonly used to identify the presence of abnormal hemoglobin variants, including hemoglobin S (HbS). Elevated levels of HbS on hemoglobin electrophoresis confirm the diagnosis of SCD.
Genetic testing plays a central role in confirming the diagnosis of SCD, particularly in cases where the clinical and laboratory findings are inconclusive. Molecular genetic testing can identify specific mutations in the HBB gene, confirming the presence of SCD.
In addition to these specific criteria, the DSM-5 emphasizes the importance of considering the patient’s clinical history, family history, and physical examination findings in the diagnostic process. Patients with SCD often present with characteristic symptoms such as chronic anemia, pain crises, jaundice, and susceptibility to infections.
Overall, the diagnosis of SCD according to DSM-5 guidelines requires a comprehensive approach that integrates clinical, laboratory, and genetic testing findings. Accurate and timely diagnosis is essential for initiating appropriate management strategies and providing comprehensive care for individuals affected by this complex genetic disorder. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
Treatment Regimens and Patient Education
Effective management of sickle cell disease (SCD) involves a combination of treatment regimens aimed at alleviating symptoms, preventing complications, and improving overall quality of life. Additionally, patient education plays a crucial role in empowering individuals with SCD to manage their condition effectively and make informed decisions about their health.
Treatment Regimens:
- Pain Management: Pain crises, or sickle cell crises, are a hallmark feature of SCD and can be managed with various pain medications, including nonsteroidal anti-inflammatory drugs (NSAIDs), opioids, and other analgesics. Intravenous fluids and heat therapy may also help alleviate pain during crises.
- Hydroxyurea Therapy: Hydroxyurea is a medication that stimulates the production of fetal hemoglobin, which has a higher affinity for oxygen and can help prevent sickling of red blood cells. Hydroxyurea has been shown to reduce the frequency of pain crises, acute chest syndrome, and hospitalizations in individuals with SCD.
- Blood Transfusions: In cases of severe anemia or complications such as acute chest syndrome or stroke, red blood cell transfusions may be necessary to improve oxygen delivery and alleviate symptoms.
- Antibiotic Prophylaxis: Due to their increased susceptibility to infections, individuals with SCD may benefit from antibiotic prophylaxis to prevent bacterial infections, particularly those caused by Streptococcus pneumoniae.
- Bone Marrow Transplantation: For select individuals with severe SCD, bone marrow transplantation may offer a potential cure by replacing the defective hematopoietic stem cells with healthy donor cells. However, this treatment option is associated with significant risks and is typically reserved for those with severe disease who have a suitable donor.
Patient Education:
- Hydration: Adequate hydration is essential for individuals with SCD to prevent dehydration, which can trigger pain crises and other complications. Patients should be educated about the importance of drinking plenty of fluids, particularly during hot weather or during physical activity.
- Infection Prevention: Patients should be educated about the increased risk of infections associated with SCD and the importance of infection prevention measures, including vaccination, regular hand hygiene, and avoiding exposure to sick individuals.
- Pain Management Strategies: Patients should be provided with information about strategies for managing pain at home, including the use of heat therapy, relaxation techniques, and proper positioning to alleviate discomfort during pain crises.
- Regular Medical Follow-Up: Regular medical follow-up is crucial for individuals with SCD to monitor their condition, assess treatment efficacy, and detect and manage complications early. Patients should be encouraged to adhere to their scheduled appointments and communicate any changes in their symptoms to their healthcare providers promptly.
- Genetic Counseling: Patients and their families should receive genetic counseling to understand the inheritance pattern of SCD and the implications for family planning. This includes discussing the risks of passing the disease to future children and available options for prenatal testing and screening.
In summary, comprehensive management of sickle cell disease involves a multifaceted approach that combines pharmacological interventions with patient education and support. Empowering individuals with SCD with knowledge about their condition and self-management strategies is essential for optimizing outcomes and improving their overall quality of life. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
Conclusion
Sickle cell disease (SCD) presents a complex challenge, affecting millions worldwide. Through examining its causes, signs and symptoms, etiology, pathophysiology, DSM-5 diagnosis, treatment regimens, and patient education, we uncover the multifaceted nature of this genetic blood disorder. Understanding the genetic mutation underlying SCD, its impact on hemoglobin production, and the resulting abnormal behavior of red blood cells is crucial for accurate diagnosis and targeted treatment approaches. Advances in treatment, including pain management, hydroxyurea therapy, and antibiotic prophylaxis, have significantly improved outcomes for those with SCD. Moreover, patient education plays a vital role in empowering individuals to manage their condition effectively, emphasizing hydration, infection prevention, pain management, and regular medical follow-up. Through ongoing research, advocacy efforts, and comprehensive care, we strive to address the challenges of SCD and improve the quality of life for affected individuals in the future. (Nursing Paper Example on Understanding Sickle Cell Disease: An Overview)
References
Do you need a similar assignment done for you from scratch? Order now!
Use Discount Code "Newclient" for a 15% Discount!